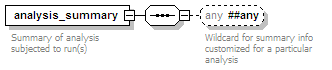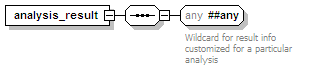| source |
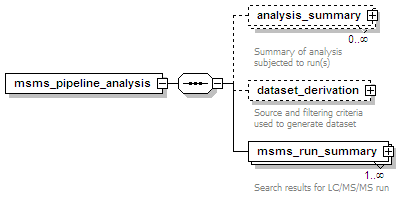
<xs:element name="msms_pipeline_analysis">
<xs:complexType>
<xs:sequence>
<xs:element name="analysis_summary" minOccurs="0" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Summary of analysis subjected to run(s)</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:sequence>
<xs:any namespace="##any" processContents="lax" minOccurs="0">
<xs:annotation>
<xs:documentation>Wildcard for summary info customized for a particular analysis</xs:documentation>
</xs:annotation>
</xs:any>
</xs:sequence>
<xs:attribute name="time" type="xs:dateTime" use="required">
<xs:annotation>
<xs:documentation>Time analysis complete (unique id)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="analysis" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Name of analysis program</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="version" type="xs:string">
<xs:annotation>
<xs:documentation>Release</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
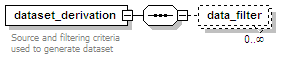
<xs:element name="dataset_derivation" minOccurs="0">
<xs:annotation>
<xs:documentation>Source and filtering criteria used to generate dataset</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:sequence>
<xs:element name="data_filter" minOccurs="0" maxOccurs="unbounded">
<xs:complexType>
<xs:attribute name="number" type="xs:nonNegativeInteger" use="required"/>
<xs:attribute name="parent_file" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>File from which derived</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="windows_parent" type="xs:string"/>
<xs:attribute name="description" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>filtering criteria applied to data</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
</xs:sequence>
<xs:attribute name="generation_no" type="xs:nonNegativeInteger" use="required">
<xs:annotation>
<xs:documentation>number preceding filter generations</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
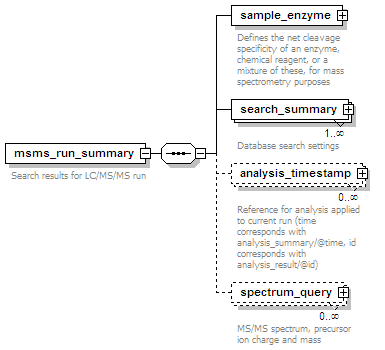
<xs:element name="msms_run_summary" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Search results for LC/MS/MS run</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:sequence>
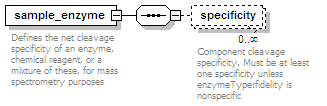
<xs:element name="sample_enzyme">
<xs:annotation>
<xs:documentation>Defines the net cleavage specificity of an enzyme, chemical reagent, or a mixture of these, for mass spectrometry purposes</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:sequence>
<xs:element name="specificity" minOccurs="0" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Component cleavage specificity. Must be at least one specificity unless enzymeType:fidelity is nonspecific </xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:attribute name="sense" use="required">
<xs:annotation>
<xs:documentation>Defines whether cleavage occurs on the C-terminal or N-terminal side of the residue(s) listed in cut</xs:documentation>
</xs:annotation>
<xs:simpleType>
<xs:restriction base="xs:string">
<xs:enumeration value="C"/>
<xs:enumeration value="N"/>
</xs:restriction>
</xs:simpleType>
</xs:attribute>
<xs:attribute name="min_spacing" type="xs:nonNegativeInteger" default="1">
<xs:annotation>
<xs:documentation>minimum separation between adjacent cleavages</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="cut" use="required">
<xs:annotation>
<xs:documentation>One or more 1-letter residue codes. Enzyme cleaves on the sense side of the residue(s) listed in cut unless one of the residues listed in no_cut is adjacent to the potential cleavage site</xs:documentation>
</xs:annotation>
<xs:simpleType>
<xs:restriction base="xs:string">
<xs:minLength value="1"/>
<xs:maxLength value="20"/>
<xs:pattern value="[A,C-I,K-N,P-T,VWY]+"/>
</xs:restriction>
</xs:simpleType>
</xs:attribute>
<xs:attribute name="no_cut" use="optional">
<xs:annotation>
<xs:documentation>Zero or more 1-letter residue codes. Enzyme cleaves on the sense side of the residue(s) listed in cut unless one of the residues listed in no_cut is adjacent to the potential cleavage site</xs:documentation>
</xs:annotation>
<xs:simpleType>
<xs:restriction base="xs:string">
<xs:minLength value="0"/>
<xs:maxLength value="20"/>
</xs:restriction>
</xs:simpleType>
</xs:attribute>
</xs:complexType>
</xs:element>
</xs:sequence>
<xs:attribute name="name" use="required">
<xs:annotation>
<xs:documentation>Controlled code name for the enzyme that can be referred to by applications</xs:documentation>
</xs:annotation>
<xs:simpleType>
<xs:restriction base="xs:string">
<xs:minLength value="1"/>
</xs:restriction>
</xs:simpleType>
</xs:attribute>
<xs:attribute name="description" type="xs:string" use="optional">
<xs:annotation>
<xs:documentation>Free text to describe alternative names, special conditions, etc.</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="fidelity" use="optional" default="specific">
<xs:annotation>
<xs:documentation>Semispecific means that at least one end of a pepide must conform to the cleavage specificity, (unless the peptide was at the terminus of the parent sequence). Nonspecific means that neither end of a peptide must conform to the cleavage specificity.</xs:documentation>
</xs:annotation>
<xs:simpleType>
<xs:restriction base="xs:string">
<xs:enumeration value="specific"/>
<xs:enumeration value="semispecific"/>
<xs:enumeration value="nonspecific"/>
</xs:restriction>
</xs:simpleType>
</xs:attribute>
<xs:attribute name="independent" type="xs:boolean" use="optional" default="1">
<xs:annotation>
<xs:documentation>If there are multiple specificities and independent is true, then a single peptide cannot exhibit one specificity at one terminus and a different specificity at the other. If independent is false, then a single peptide can exhibit mixed specificities.</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
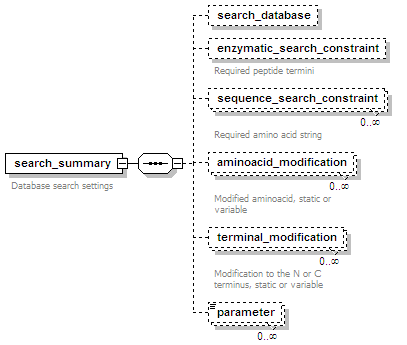
<xs:element name="search_summary" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Database search settings</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:sequence>
<xs:element name="search_database" minOccurs="0">
<xs:complexType>
<xs:attribute name="local_path" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Full path address of database on local computer</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="URL" type="xs:string"/>
<xs:attribute name="database_name" type="xs:string"/>
<xs:attribute name="orig_database_url" type="xs:string"/>
<xs:attribute name="database_release_date" type="xs:dateTime"/>
<xs:attribute name="database_release_identifier" type="xs:string"/>
<xs:attribute name="size_in_db_entries" type="xs:nonNegativeInteger"/>
<xs:attribute name="size_of_residues" type="xs:nonNegativeInteger"/>
<xs:attribute name="type" use="required">
<xs:annotation>
<xs:documentation>Database type (AA=amino acid, NA=nucleic acid)</xs:documentation>
</xs:annotation>
<xs:simpleType>
<xs:restriction base="xs:string">
<xs:enumeration value="AA"/>
<xs:enumeration value="NA"/>
</xs:restriction>
</xs:simpleType>
</xs:attribute>
</xs:complexType>
</xs:element>
<xs:element name="enzymatic_search_constraint" minOccurs="0">
<xs:annotation>
<xs:documentation>Required peptide termini</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:attribute name="enzyme" type="xs:string" use="required"/>
<xs:attribute name="max_num_internal_cleavages" type="xs:nonNegativeInteger" use="required">
<xs:annotation>
<xs:documentation>Maximum number of enzyme cleavage sites allowable within peptide</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="min_number_termini" type="xs:nonNegativeInteger" use="required">
<xs:annotation>
<xs:documentation>Minimum number of termini compatible with enzymatic cleavage</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
<xs:element name="sequence_search_constraint" minOccurs="0" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Required amino acid string</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:attribute name="sequence" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Required amino acid string</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
<xs:element name="aminoacid_modification" minOccurs="0" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Modified aminoacid, static or variable</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:attribute name="aminoacid" type="xs:string" use="required"/>
<xs:attribute name="massdiff" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Mass difference with respect to unmodified aminoacid, must begin with either + (nonnegative) or - [e.g. +1.05446 or -2.3342]</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="mass" type="xs:float" use="required">
<xs:annotation>
<xs:documentation>Mass of modified aminoacid</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="variable" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Y if both modified and unmodified aminoacid could be present in the dataset, N if only modified aminoacid can be present</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="peptide_terminus" type="xs:string">
<xs:annotation>
<xs:documentation>whether modification can reside only at protein terminus (specified 'n', 'c', or 'nc')</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="symbol" type="aa_symbolType">
<xs:annotation>
<xs:documentation>Special symbol used by search engine to designate this modification</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="binary" type="xs:string">
<xs:annotation>
<xs:documentation>Y if each peptide must have only modified or unmodified aminoacid, N if a peptide may contain both modified and unmodified aminoacid</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="description" type="xs:string"/>
</xs:complexType>
</xs:element>
<xs:element name="terminal_modification" minOccurs="0" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Modification to the N or C terminus, static or variable</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:attribute name="terminus" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>n for N-terminus, c for C-terminus</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="massdiff" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Mass difference with respect to unmodified terminus</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="mass" type="xs:float" use="required">
<xs:annotation>
<xs:documentation>Mass of modified terminus</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="variable" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Y if both modified and unmodified terminus could be present in the dataset, N if only modified terminus can be present</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="symbol" type="term_symbolType">
<xs:annotation>
<xs:documentation>Special symbol used by search engine to designate this modification</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="protein_terminus" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>whether modification can reside only at protein terminus (specified n or c)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="description" type="xs:string"/>
</xs:complexType>
</xs:element>
<xs:element name="parameter" type="nameValueType" minOccurs="0" maxOccurs="unbounded"/>
</xs:sequence>
<xs:attribute name="base_name" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Full path location of mzXML file for this search run (without the .mzXML extension)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="search_engine" type="engineType" use="required">
<xs:annotation>
<xs:documentation>SEQUEST, Mascot, COMET, etc</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="precursor_mass_type" type="massType" use="required">
<xs:annotation>
<xs:documentation>average or monoisotopic</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="fragment_mass_type" type="massType" use="required">
<xs:annotation>
<xs:documentation>average or monoisotopic</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="out_data_type" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Format of file storing the runner up peptides (if not present in pepXML)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="out_data" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>runner up search hit data type extension (e.g. .tgz)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="search_id" type="positiveInt" use="required">
<xs:annotation>
<xs:documentation>matches id in search hit</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
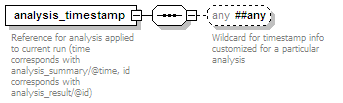
<xs:element name="analysis_timestamp" minOccurs="0" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Reference for analysis applied to current run (time corresponds with analysis_summary/@time, id corresponds with analysis_result/@id)</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:sequence>
<xs:any namespace="##any" processContents="lax" minOccurs="0">
<xs:annotation>
<xs:documentation>Wildcard for timestamp info customized for a particular analysis</xs:documentation>
</xs:annotation>
</xs:any>
</xs:sequence>
<xs:attribute name="time" type="xs:dateTime" use="required">
<xs:annotation>
<xs:documentation>Date of analysis</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="analysis" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Analysis name</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="id" type="positiveInt" use="required">
<xs:annotation>
<xs:documentation>Unique identifier for each type of analysis</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
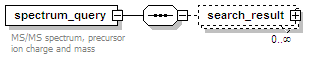
<xs:element name="spectrum_query" minOccurs="0" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>MS/MS spectrum, precursor ion charge and mass</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:sequence>
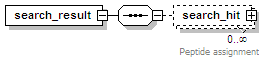
<xs:element name="search_result" minOccurs="0" maxOccurs="unbounded">
<xs:complexType>
<xs:sequence>
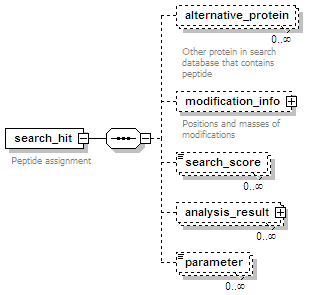
<xs:element name="search_hit" minOccurs="0" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Peptide assignment</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:sequence>
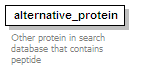
<xs:element name="alternative_protein" minOccurs="0" maxOccurs="unbounded">
<xs:annotation>
<xs:documentation>Other protein in search database that contains peptide</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:attribute name="protein" type="xs:string" use="required"/>
<xs:attribute name="protein_descr" type="xs:string"/>
<xs:attribute name="num_tol_term" type="xs:nonNegativeInteger"/>
<xs:attribute name="protein_mw" type="xs:double"/>
</xs:complexType>
</xs:element>
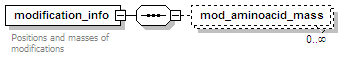
<xs:element name="modification_info" minOccurs="0">
<xs:annotation>
<xs:documentation>Positions and masses of modifications</xs:documentation>
</xs:annotation>
<xs:complexType>
<xs:sequence>
<xs:element name="mod_aminoacid_mass" minOccurs="0" maxOccurs="unbounded">
<xs:complexType>
<xs:attribute name="position" type="xs:nonNegativeInteger" use="required">
<xs:annotation>
<xs:documentation>modified aminoacid position in peptide [ranging from 1 to peptide length]</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="mass" type="xs:double" use="required">
<xs:annotation>
<xs:documentation>modified mass of aminoacid</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
</xs:sequence>
<xs:attribute name="mod_nterm_mass" type="xs:double">
<xs:annotation>
<xs:documentation>Mass of modified N terminus</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="mod_cterm_mass" type="xs:double">
<xs:annotation>
<xs:documentation>Mass of modified C terminus</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="modified_peptide" type="xs:string">
<xs:annotation>
<xs:documentation>Peptide sequence (with indicated modifications)</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
<xs:element name="search_score" type="nameValueType" minOccurs="0" maxOccurs="unbounded"/>
<xs:element name="analysis_result" minOccurs="0" maxOccurs="unbounded">
<xs:complexType>
<xs:sequence>
<xs:any namespace="##any" processContents="lax">
<xs:annotation>
<xs:documentation>Wildcard for result info customized for a particular analysis</xs:documentation>
</xs:annotation>
</xs:any>
</xs:sequence>
<xs:attribute name="analysis" type="xs:string" use="required"/>
<xs:attribute name="id" type="positiveInt" default="1"/>
</xs:complexType>
</xs:element>
<xs:element name="parameter" type="nameValueType" minOccurs="0" maxOccurs="unbounded"/>
</xs:sequence>
<xs:attribute name="hit_rank" type="positiveInt" use="required"/>
<xs:attribute name="peptide" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Peptide aminoacid sequence (with no indicated modifications)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="peptide_prev_aa" type="xs:string">
<xs:annotation>
<xs:documentation>Aminoacid preceding peptide (- if none)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="peptide_next_aa" type="xs:string">
<xs:annotation>
<xs:documentation>Aminoacid following peptide (- if none)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="protein" type="xs:string" use="required"/>
<xs:attribute name="num_tot_proteins" type="xs:unsignedInt" use="required">
<xs:annotation>
<xs:documentation>Number of unique proteins in search database containing peptide</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="num_matched_ions" type="xs:nonNegativeInteger">
<xs:annotation>
<xs:documentation>Number of peptide fragment ions found in spectrum</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="tot_num_ions" type="xs:nonNegativeInteger">
<xs:annotation>
<xs:documentation>Number of peptide fragment ions predicted for peptide</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="calc_neutral_pep_mass" type="xs:float" use="required"/>
<xs:attribute name="massdiff" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Mass(precursor ion) - Mass(peptide)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="num_tol_term" type="xs:nonNegativeInteger">
<xs:annotation>
<xs:documentation>Number of peptide termini consistent with cleavage by sample enzyme</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="num_missed_cleavages" type="xs:integer">
<xs:annotation>
<xs:documentation>Number of sample enzyme cleavage sites internal to peptide</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="is_rejected" default="0">
<xs:annotation>
<xs:documentation>Potential use in future for user manual validation (0 or 1)</xs:documentation>
</xs:annotation>
<xs:simpleType>
<xs:restriction base="xs:nonNegativeInteger">
<xs:enumeration value="0"/>
<xs:enumeration value="1"/>
</xs:restriction>
</xs:simpleType>
</xs:attribute>
<xs:attribute name="protein_descr" type="xs:string">
<xs:annotation>
<xs:documentation>Extracted from search database</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="calc_pI" type="xs:string"/>
<xs:attribute name="protein_mw" type="xs:double"/>
</xs:complexType>
<xs:unique name="unique_result_analysis_id">
<xs:annotation>
<xs:documentation>analysis and id must be unique within a search_hit</xs:documentation>
</xs:annotation>
<xs:selector xpath="./pepx:analysis_result"/>
<xs:field xpath="@analysis"/>
<xs:field xpath="@id"/>
</xs:unique>
</xs:element>
</xs:sequence>
<xs:attribute name="search_id" type="positiveInt" default="1">
<xs:annotation>
<xs:documentation>Unique identifier to search summary</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
</xs:element>
<!-- search result -->
</xs:sequence>
<xs:attribute name="spectrum" type="xs:string" use="required"/>
<xs:attribute name="start_scan" type="xs:unsignedInt" use="required">
<xs:annotation>
<xs:documentation>first scan number integrated into MS/MS spectrum</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="end_scan" type="xs:unsignedInt" use="required">
<xs:annotation>
<xs:documentation>last scan number integrated into MS/MS spectrum</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="precursor_neutral_mass" type="xs:float" use="required"/>
<xs:attribute name="assumed_charge" type="xs:nonNegativeInteger" use="required">
<xs:annotation>
<xs:documentation>Precursor ion charge used for search</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="search_specification" type="xs:string">
<xs:annotation>
<xs:documentation>Search constraint applied specifically to this query</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="index" type="positiveInt" use="required">
<xs:annotation>
<xs:documentation>Unique identifier</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
<xs:unique name="unique_search_id">
<xs:annotation>
<xs:documentation>search_id must be unique within each msms_run_summary</xs:documentation>
</xs:annotation>
<xs:selector xpath="./pepx:search_result"/>
<xs:field xpath="@search_id"/>
</xs:unique>
</xs:element>
</xs:sequence>
<xs:attribute name="base_name" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>full path file name of mzXML (minus the .mzXML)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="raw_data_type" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>raw data type extension (e.g. .mzXML)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="raw_data" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>raw data type extension (e.g. .mzXML)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="msManufacturer" type="xs:string">
<xs:annotation>
<xs:documentation>Manufacturer of MS/MS instrument</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="msModel" type="xs:string">
<xs:annotation>
<xs:documentation>Instrument model (cf mzXML)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="msIonization" type="xs:string">
<xs:annotation>
<xs:documentation>Instrument model (cf mzXML)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="msMassAnalyzer" type="xs:string">
<xs:annotation>
<xs:documentation>Ion trap, etc (cf mzXML)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="msDetector" type="xs:string">
<xs:annotation>
<xs:documentation>EMT, etc(cf mzXML)</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
<xs:unique name="unique_timestamp_analysis_time">
<xs:annotation>
<xs:documentation>analysis_timestamp analysis and time must be unique within each msms_run_summary</xs:documentation>
</xs:annotation>
<xs:selector xpath="./pepx:analysis_timestamp"/>
<xs:field xpath="@analysis"/>
<xs:field xpath="@time"/>
</xs:unique>
<xs:key name="search_summary_id">
<xs:selector xpath="./pepx:search_summary"/>
<xs:field xpath="@search_id"/>
</xs:key>
<xs:keyref name="search_result_id" refer="search_summary_id">
<xs:annotation>
<xs:documentation>search_id within each search_hit must correspond with that of a search_summary</xs:documentation>
</xs:annotation>
<xs:selector xpath="./pepx:spectrum_query/pepx:search_result"/>
<xs:field xpath="@search_id"/>
</xs:keyref>
<xs:key name="timestamp_analysis_id">
<xs:selector xpath="./pepx:analysis_timestamp"/>
<xs:field xpath="@analysis"/>
<xs:field xpath="@id"/>
</xs:key>
<xs:keyref name="result_analysis_id" refer="timestamp_analysis_id">
<xs:annotation>
<xs:documentation>analysis and id in analysis_result must correspond with those in an analysis_timestamp element</xs:documentation>
</xs:annotation>
<xs:selector xpath="./pepx:spectrum_query/pepx:search_result/pepx:search_hit/pepx:analysis_result"/>
<xs:field xpath="@analysis"/>
<xs:field xpath="@id"/>
</xs:keyref>
</xs:element>
</xs:sequence>
<xs:attribute name="name" type="xs:string">
<xs:annotation>
<xs:documentation>Summary name (currently not used)</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="date" type="xs:dateTime" use="required">
<xs:annotation>
<xs:documentation>Date pepXML file was written</xs:documentation>
</xs:annotation>
</xs:attribute>
<xs:attribute name="summary_xml" type="xs:string" use="required">
<xs:annotation>
<xs:documentation>Full path self reference</xs:documentation>
</xs:annotation>
</xs:attribute>
</xs:complexType>
<xs:key name="summary_analysis_time">
<xs:selector xpath="./pepx:analysis_summary"/>
<xs:field xpath="@time"/>
<xs:field xpath="@analysis"/>
</xs:key>
<xs:keyref name="timestamp_analysis_time" refer="summary_analysis_time">
<xs:annotation>
<xs:documentation>time and analysis within timestamp must correspond with those within analysis_summary element</xs:documentation>
</xs:annotation>
<xs:selector xpath="./pepx:msms_run_summary/pepx:analysis_timestamp"/>
<xs:field xpath="@time"/>
<xs:field xpath="@analysis"/>
</xs:keyref>
<xs:unique name="unique_search_summary_basename">
<xs:annotation>
<xs:documentation>search_summary base_name (mzXML file) must be unique within document</xs:documentation>
</xs:annotation>
<xs:selector xpath="./pepx:msms_run_summary/pepx:search_summary"/>
<xs:field xpath="@base_name"/>
</xs:unique>
<xs:unique name="unique_query_index">
<xs:annotation>
<xs:documentation>spectrum_query index must be unique in document</xs:documentation>
</xs:annotation>
<xs:selector xpath="./pepx:msms_run_summary/pepx:spectrum_query"/>
<xs:field xpath="@index"/>
</xs:unique>
</xs:element> |